
Sickle Beta Thalassemia
Rare Disease Sickle Beta Thalassemia|RMCH.

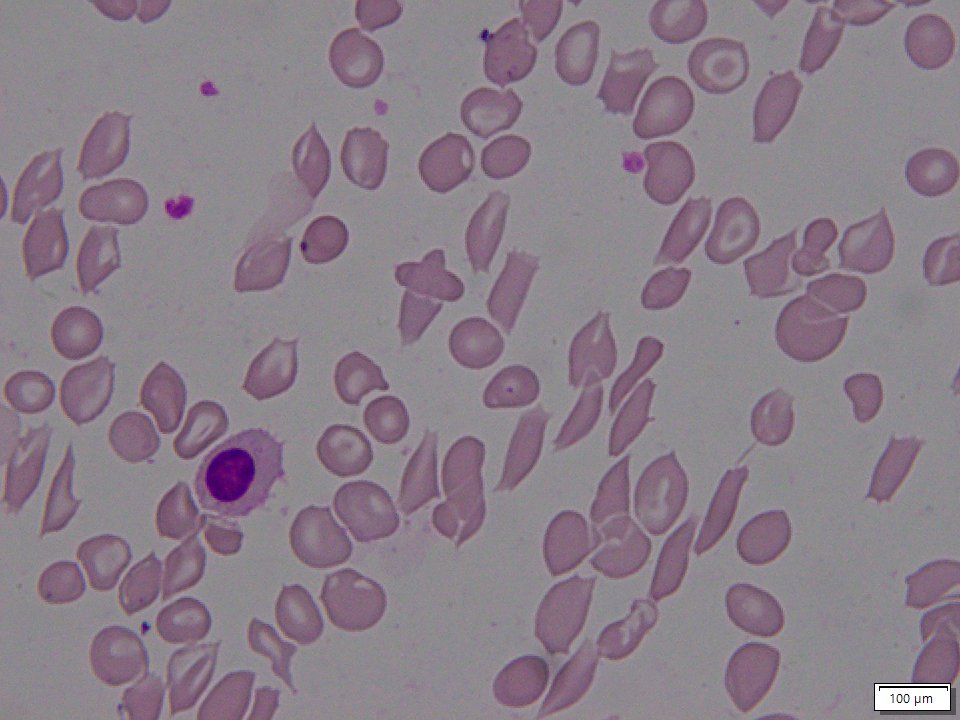
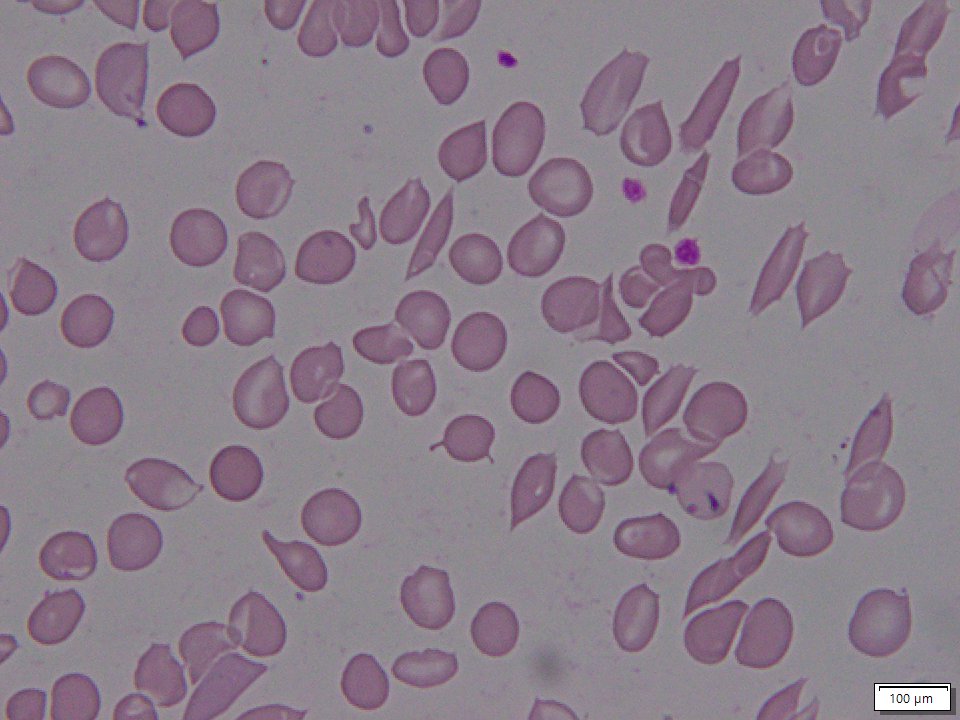
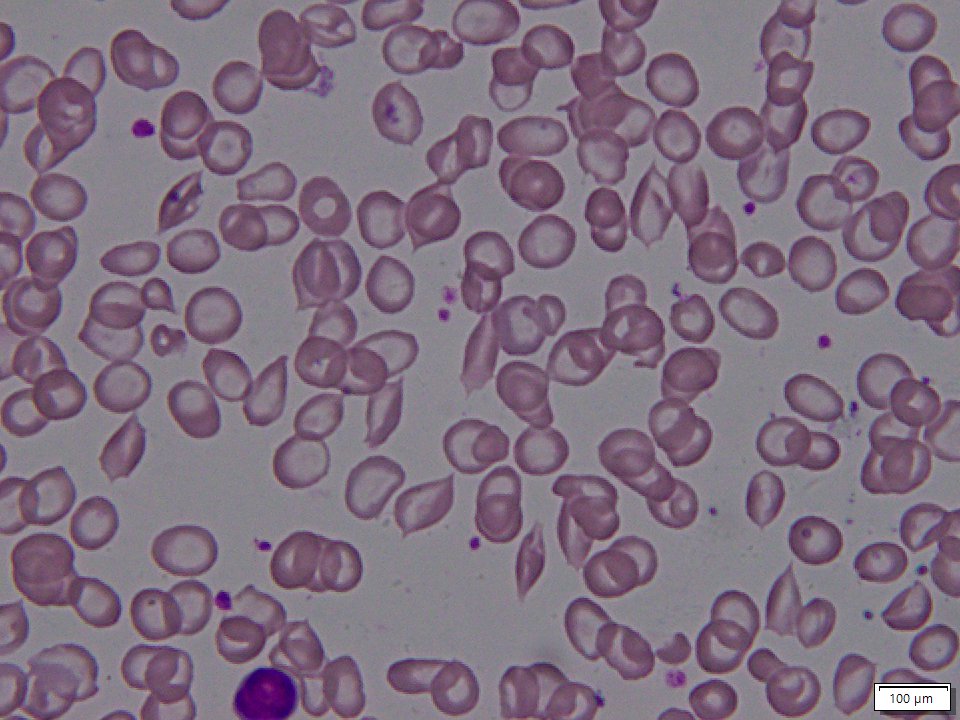
NIH GARD Information: Sickle beta thalassemia
This information is provided by the National Institutes of Health (NIH) Genetic and Rare Diseases Information Center (GARD).
Synonyms
- Hemoglobin sickle-beta thalassemia
- Hb S beta-thalassemia
- Sickle cell – beta-thalassemia disease
- HbS-beta-thalassemia syndrome
- Sickle cell-beta-thalassemia disease syndrome
- HbS – beta-thalassemia
Overview
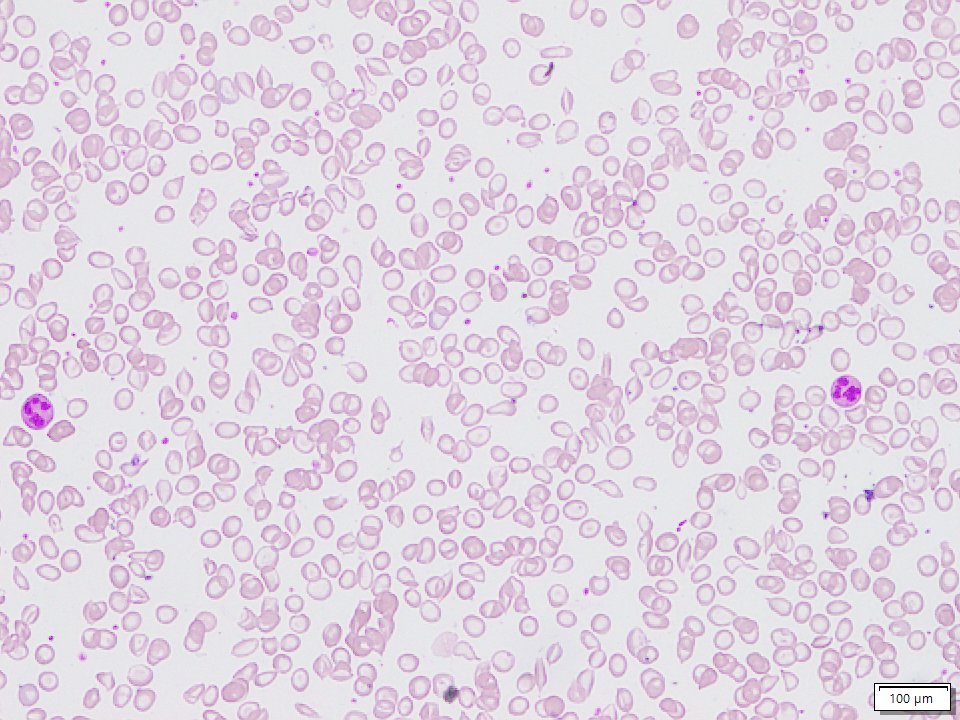
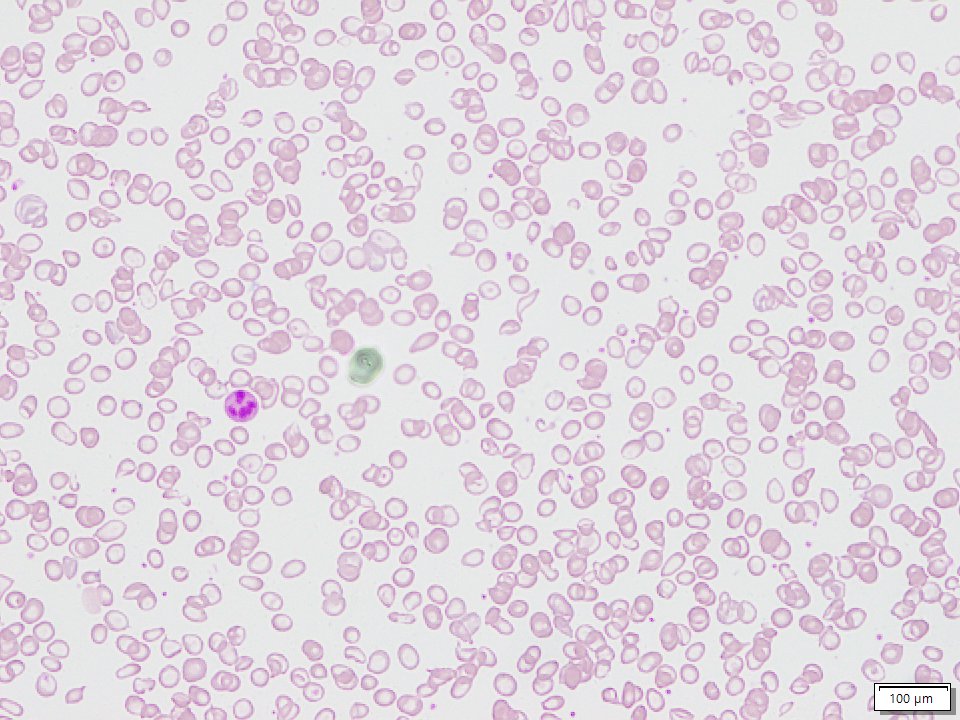
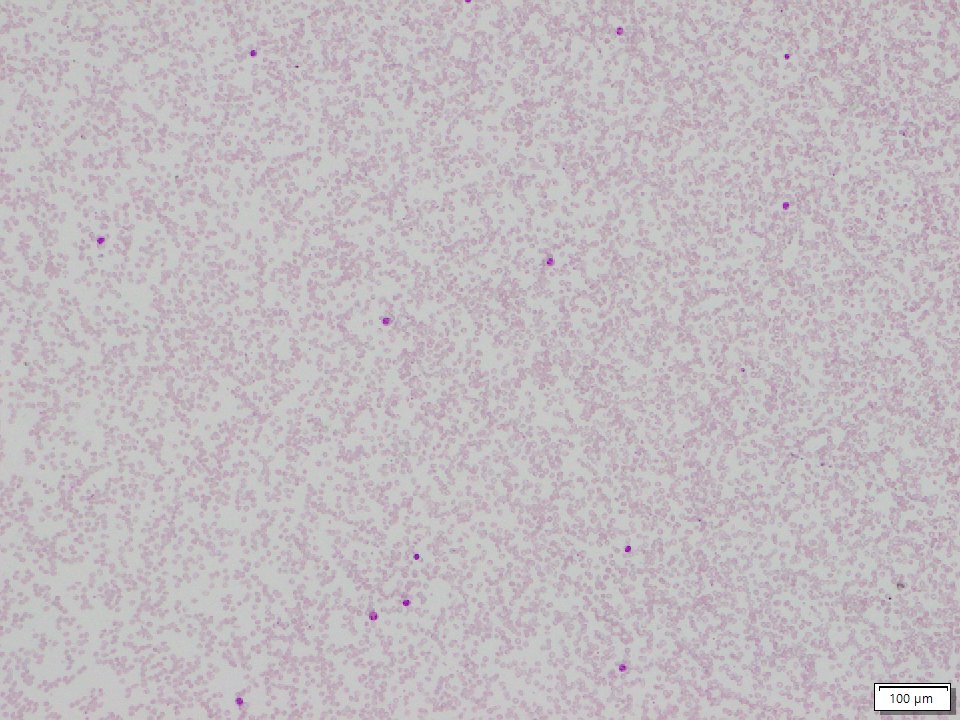
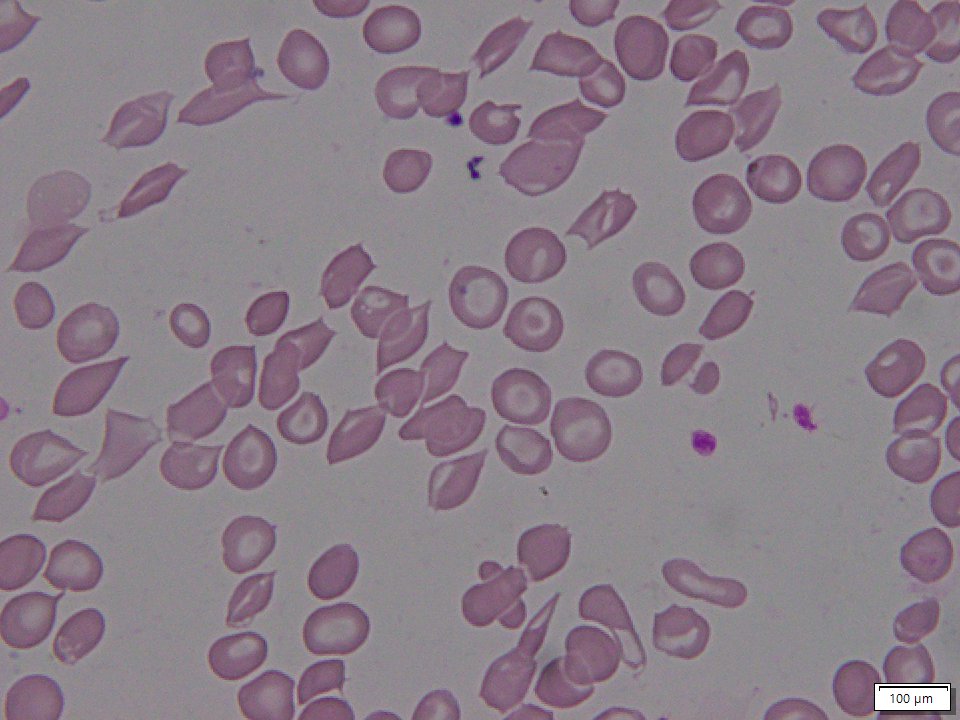
Sickle beta thalassemia is an inherited condition that affects hemoglobin, the protein in red blood cells that carries oxygen to different parts of the body. It is a type of sickle cell disease. Affected people have a different change (mutation) in each copy of their HBB gene: one that causes red blood cells to form a “sickle” or crescent shape and a second that is associated with beta thalassemia, a blood disorder that reduces the production of hemoglobin. Depending on the beta thalassemia mutation, people may have no normal hemoglobin (called sickle beta zero thalassemia) or a reduced amount of normal hemoglobin (called sickle beta plus thalassemia). The presence of sickle-shaped red blood cells, which often breakdown prematurely and can get stuck in blood vessels, combined with the reduction or absence of mature red blood cells leads to the many signs and symptoms of sickle beta thalassemia. Features, which may include anemia (low levels of red blood cells), repeated infections, and frequent episodes of pain, generally develop in early childhood and vary in severity depending on the amount of normal hemoglobin made. Sickle beta thalassemia is inherited in an autosomal recessive manner. Treatment is supportive and depends on the signs and symptoms present in each person.[8020][8021][8022]
For more information, visit GARD.